
This example uses VMD to set up a classical forcefield simulation in CP2K. You can download VMD from here, but it may well already be installed on a machine you can access.
When VMD is running you can download a protein structure using the “extensions → data → PDB database query” dropdown menus of the main VMD window. This allows a PDB structure file to be downloaded (assumes that you have an internet connection on the machine running VMD). You should get a window like
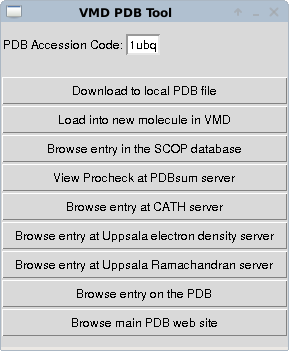
Enter the 4 character accession code of a protein, here I've put in ubiquitin - 1ubq - and select load into a new molecule. You should now see a line representation of the protein in the vmd window. The important step is now to generate a topology for this molecule that defines how it is connected and will define how interactions from the main forcefield we will use will be mapped to the atoms in the molecule we have just downloaded. To do this we can use the autopsf plugin. We select this from the “extensions → modelling → automatic PSF generator” dropdowns of the VMD main window.
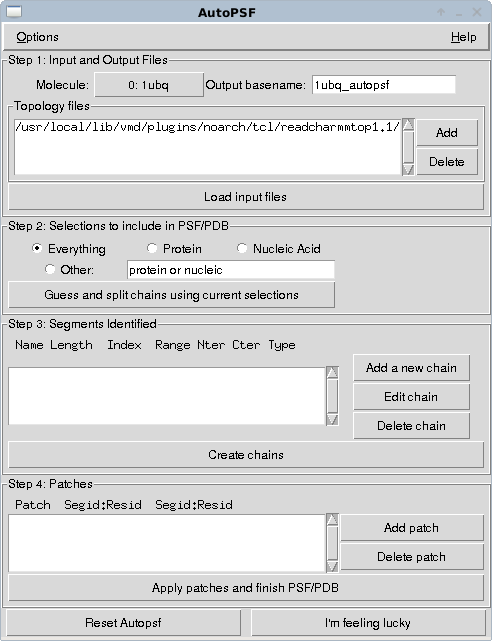
Hit I'm feeling lucky. You should now have three new files in the directory where you started VMD (or maybe where it is installed in windows if you haven't changed directory…) 1ubq_autopsf.psf, 1ubq_autopsf.pdb, 1ubq_autopsf.log.
We can easily solvate this in VMD using the “extensions → modelling → Add Solvation Box” dropdowns. Maybe it is a good idea to specify the box size you want, or as I've down a bit of padding to stop the periodic images of the protein getting too close, then hit solvate.
{{http://www.cp2k.org/_media/exercises:2015_cecam_tutorial:vmd_solvate.png}}
Now you should have solvate.psf and solvate.pdb in your directory, which contain the coordinates and connectivity information of the solvated protein. Open the .psf file edit the first line to simple read PSF. For some proteins you might now need to add ions into the solvent to neutralize the system, but we're OK with ubiquitin.
At this point we're nearly ready to run CP2K. First we need to get hold of the forcefield files. The CHARMM forcefields are distributed from the MacKrell group website charmm_ff.shtml. Download toppar_c31b1.tar.gz, extract it with something like tar -zxvf toppar_c31b1.tar.gz.
OK, finally ready to go. The first thing to do is to carry out a rough minimization to remove any close contacts between the protein and the waters that we added in VMD. These clashes would probably make molecular dynamics unstable if we just started running.
&FORCE_EVAL
METHOD FIST
&MM
&FORCEFIELD
#this assumes that you've downloaded and extracted the file as instructed
parm_file_name toppar/par_all27_prot_lipid.prm
parmtype CHM
&SPLINE
EMAX_SPLINE 1000000
RCUT_NB 10.0
&END
&END FORCEFIELD
&POISSON
&EWALD
EWALD_TYPE spme
ALPHA .44
GMAX 36
O_SPLINE 6
&END EWALD
&END POISSON
&END MM
&SUBSYS
&CELL
ABC 40.685 42.380 46.291
&END CELL
&TOPOLOGY
COORD_FILE_NAME solvate.pdb
COORDINATE pdb
CONN_FILE_FORMAT psf
CONN_FILE_NAME solvate.psf
&END TOPOLOGY
&END SUBSYS
&END FORCE_EVAL
&GLOBAL
PROJECT ubiquitin_mini
RUN_TYPE GEO_OPT
&END GLOBAL
&MOTION
&GEO_OPT
MINIMIZER LBFGS
#just do 100 steps,
#we only want to remove bad contacts
MAX_ITER 100
&END
&END MOTION
Now we can start to equilibriate the system in MD. Make a new input file as follows, and run it in the same directory (or tell it where the restart file from the minimization is)
&FORCE_EVAL
METHOD FIST
&MM
&FORCEFIELD
parm_file_name toppar/par_all27_prot_lipid.prm
parmtype CHM
&SPLINE
EMAX_SPLINE 1000000
RCUT_NB 10.0
&END
&END FORCEFIELD
&POISSON
&EWALD
EWALD_TYPE spme
ALPHA .44
GMAX 36
O_SPLINE 6
&END EWALD
&END POISSON
&END MM
&SUBSYS
&CELL
ABC 40.685 42.380 46.291
&END CELL
&TOPOLOGY
COORD_FILE_NAME solvate.pdb
COORDINATE pdb
CONN_FILE_FORMAT psf
CONN_FILE_NAME solvate.psf
&END TOPOLOGY
&PRINT
&TOPOLOGY_INFO
PSF_INFO
&END
&END
&END SUBSYS
&END FORCE_EVAL
&GLOBAL
PROJECT ubiquitin_md
RUN_TYPE MD
&END GLOBAL
&EXT_RESTART
RESTART_FILE_NAME ubiquitin_mini-1.restart
RESTART_DEFAULT F
RESTART_POS T
&END EXT_RESTART
&MOTION
&MD
ENSEMBLE NVT
STEPS 1000
TIMESTEP 0.5
TEMPERATURE 298
&THERMOSTAT
REGION MASSIVE
TYPE CSVR
&CSVR
TIMECON 100.
&END
&END
&END MD
&END MOTION
We take the coordinates from previous minimization run using the external restart option in CP2K. Hopefully this works for you and you can explore the MD facilities in the code. You'd need to equilibriate for quite some ns before starting production, maybe including some NPT to make sure the box size is correct.
(This exercise is heavily based on material I-Feng William Kuo (Lawrence Livermore National Laboratory) presented at previous CP2K workshops)