Table of Contents
Nanostructures and adsorption on metallic surfaces
Problem: compute activation barrier for the last step (6 to 7) of the cyclodehydrogenation reaction CHP@Cu(111) → TBC
- Original author: Carlo Pignedoli
- Complete source and output files: NEB.tar.xz
Introduction
The system is composed by a molecule (56 atoms) adsorbed on a Cu(111) slab (~2900 atoms). The molecule is treated within DFT (PM61) in the exercise) and the substrate with EAM2) potentials. The substrate and the molecule are coupled through a par potential mimicking the van der Waals interaction and Pauli repulsion.3)
Exercise 1: NEB
We need MULTIPLE_FORCE_EVALS:
- In a first section we define how to partition the whole system in a DFT+EMPIRICAL part
- in a second section we define the force fields for the EMPIRICAL part
- and in the third section we define the parameters for the DFT part.
- The fourth and final section defines the target of the calculation(GEO_OPT, NEB, Molecular dynamics..)
The files for this part of the exercise are found in the EX2 folder.
First Section
Here we specify that our system consists of two fragments: atoms 1 to 56 and atoms 57 to 2936,
the whole system is contained in the cell specified in FORCE_EVAL/SUBSYS section.
We declare that the first FORCE_EVAL section following the present one will be dedicated to FRAGMENTS 1 and 2 (it will contain the empirical potentials)
and the second FORCE_EVAL section will be devoted to FRAGMENT 1 only (the DFT treatment of the molecule).
&MULTIPLE_FORCE_EVALS
FORCE_EVAL_ORDER 2 3
MULTIPLE_SUBSYS T
&END
&FORCE_EVAL
METHOD MIXED
&MIXED
MIXING_TYPE GENMIX
GROUP_PARTITION 1 1
&GENERIC
ERROR_LIMIT 1.0E-10
MIXING_FUNCTION E1+E2
VARIABLES E1 E2
&END
&MAPPING
&FORCE_EVAL_MIXED
&FRAGMENT 1
1 56
&END
&FRAGMENT 2
57 2936
&END
&END
&FORCE_EVAL 1
DEFINE_FRAGMENTS 1 2
&END
&FORCE_EVAL 2
DEFINE_FRAGMENTS 1
&END
&END
&END
&SUBSYS
&COLVAR
&DISTANCE
ATOMS 49 50
&END
&END
&CELL
ABC 40.764229 39.715716 70.
&END CELL
&TOPOLOGY
COORD_FILE_NAME ./s.xyz
COORDINATE XYZ
CONNECTIVITY OFF
&END TOPOLOGY
&END SUBSYS
&END
Second Section
In this section we define the empirical potentials for the whole system:
EAM will be used for the Cu-Cu interactions (parametrized in the file Files/CU.pot),
a C6/R^6 + pauli repulsion will be defined for C-Cu and H-Cu and, finally,
NULL interactions will be defined for C-C, C-H and H-H since these will be obtained from DFT in the next section.
The charge of every specie (zero in our example) as well as the interaction within each possible type-pair has to be defined.
&FORCE_EVAL
METHOD FIST
&MM
&FORCEFIELD
&SPLINE
EPS_SPLINE 1.0E-6
EMAX_SPLINE 0.9
&END
&CHARGE
ATOM Cu
CHARGE 0.0
&END CHARGE
&CHARGE
ATOM H
CHARGE 0.0
&END CHARGE
&CHARGE
ATOM C
CHARGE 0.0
&END CHARGE
&NONBONDED
&GENPOT
atoms Cu C
FUNCTION A*exp(-av*r)+B*exp(-ac*r)-C/(r^6)
VARIABLES r
PARAMETERS A av B ac C
VALUES 4.13643 1.33747 115.82004 2.206825 75.40708524085266692113
RCUT 15
&END GENPOT
&GENPOT
atoms Cu H
FUNCTION A*exp(-av*r)+B*exp(-ac*r)-C/(r^6)
VARIABLES r
PARAMETERS A av B ac C
VALUES 0.878363 1.33747 24.594164 2.206825 21.32834305207502214234
RCUT 15
&END GENPOT
&LENNARD-JONES
atoms C H
EPSILON 0.0
SIGMA 3.166
RCUT 15
&END LENNARD-JONES
&LENNARD-JONES
atoms H H
EPSILON 0.0
SIGMA 3.166
RCUT 15
&END LENNARD-JONES
&LENNARD-JONES
atoms C C
EPSILON 0.0
SIGMA 3.166
RCUT 15
&END LENNARD-JONES
&EAM
atoms Cu Cu
PARM_FILE_NAME ../../Files/CU.pot
&END EAM
&END NONBONDED
&END FORCEFIELD
&POISSON
&EWALD
EWALD_TYPE none
&END EWALD
&END POISSON
&END
&SUBSYS
&CELL
ABC 40.764229 39.715716 70.
&END CELL
&TOPOLOGY
COORD_FILE_NAME ./s.xyz
COORDINATE XYZ
CONNECTIVITY OFF
&END TOPOLOGY
&END SUBSYS
&END
Third Section
In this section we specify the parameters for the DFT treatment of the molecule. Note that the simulation cell here is reduced with comparison to the simulation cell of the whole system.
&FORCE_EVAL
METHOD Quickstep
&DFT
&QS
METHOD PM6
EXTRAPOLATION USE_GUESS
&SE
&COULOMB
CUTOFF [angstrom] 20.0
&END
&EXCHANGE
CUTOFF [angstrom] 20.0
&END
&END
&END QS
&SCF
MAX_SCF 20
SCF_GUESS RESTART
EPS_SCF 1.0E-6
&OT
PRECONDITIONER FULL_SINGLE_INVERSE
MINIMIZER DIIS
N_DIIS 7
&END
&OUTER_SCF
MAX_SCF 10
EPS_SCF 1.0E-6
&END
&PRINT
&RESTART
&EACH
QS_SCF 0
&END
ADD_LAST NUMERIC
&END
&RESTART_HISTORY OFF
&END
&END
&END SCF
&POISSON
PERIODIC NONE
POISSON_SOLVER WAVELET
&WAVELET
SCF_TYPE 60
&END
&END POISSON
&XC
&XC_FUNCTIONAL BLYP
&END XC_FUNCTIONAL
&END XC
! &PRINT
! &MO_CUBES
! NHOMO 15
! NLUMO 15
! WRITE_CUBE T
! LOG_PRINT_KEY
! &END
! &END
&END DFT
&SUBSYS
&CELL
ABC 30 30 30
PERIODIC NONE
&END CELL
&TOPOLOGY
COORD_FILE_NAME ./f.xyz
COORDINATE xyz
&CENTER_COORDINATES
&END
&END
# &KIND H
# BASIS_SET TZV2P-MOLOPT-GTH
# POTENTIAL GTH-BLYP-q1
# &END KIND
# &KIND C
# BASIS_SET TZV2P-MOLOPT-GTH
# POTENTIAL GTH-BLYP-q4
# &END KIND
&END SUBSYS
&END FORCE_EVAL
Fourth Section
In this section we define the target of the simulation as a geometry optimization this will be MD, or BAND in other examples.
&GLOBAL
PRINT_LEVEL LOW
PROJECT INI
RUN_TYPE GEO_OPT
WALLTIME 28000
&END GLOBAL
&MOTION
&GEO_OPT
MAX_FORCE 0.0001
MAX_ITER 500
OPTIMIZER LBFGS
&LBFGS
&END
&END
&END
!&EXT_RESTART
! RESTART_FILE_NAME constr_3cc_2h_41_dft-1.restart
!&END
Tasks
- First of all obtain the equilibrium geometry for the initial and the final states (in the directories
INIandFINyou will find the input files (INI/geo.inp/FIN/geo.inp) the output files (INI/out/FIN/out) and the trajectory of the geometry optimization (INI-pos-1.xyzandFIN-pos-1.xyz).- Please note that the geometry optimization of the final state is constrained with respect to the position of the final H2 molecule.
- Perform a NEB simulation to find a saddle point between
ini_eq.xyzandfin_eq.xzy.- A linear guess for the path is not a good idea in this case; a clever guess can be obtained from a series of constrained geometry optimizations where the H-H distance is forced to vary from its initial value (more than 3 Å) to the H2 equilibrium distance. Such a guess is given, in the directory
EX2, with the filesini_eq.xyz,due.xyz,tre.xyz,…,tredici.xyz,fin_eq.xyz).
Check that the initial step of the NEB calculation is reasonable and determine the activation barrier at the and of the optimization. In the directoryEX2you will find the input for the calculation (neb.inp), the output (out) and the optimization trajectory of each image of the system (NEB-pos-Replica_nr_XX-1.xyz, 14 images are used in the example)
The last section of the input described above is modified as follows:
&GLOBAL
PRINT_LEVEL LOW
PROJECT NEB
RUN_TYPE BAND
WALLTIME 28000
&END GLOBAL
&MOTION
&BAND
NPROC_REP 2
BAND_TYPE CI-NEB
NUMBER_OF_REPLICA 14
K_SPRING 0.05
&CONVERGENCE_CONTROL
MAX_FORCE 0.0030
RMS_FORCE 0.0050
MAX_DR 0.002
RMS_DR 0.005
&END
ROTATE_FRAMES F
ALIGN_FRAMES F
&CI_NEB
NSTEPS_IT 2
&END
&OPTIMIZE_BAND
OPT_TYPE DIIS
OPTIMIZE_END_POINTS F
&DIIS
MAX_STEPS 1000
&END
&END
#
&REPLICA
COORD_FILE_NAME ini_eq.xyz
&END
&REPLICA
COORD_FILE_NAME due.xyz
&END
.
.
.
.
.
&REPLICA
COORD_FILE_NAME fin_eq.xyz
&END
&PROGRAM_RUN_INFO
INITIAL_CONFIGURATION_INFO
&END
&CONVERGENCE_INFO
&END
&END
&END
We declare that we need 14 replicas and each replica has to be computed with 2 CPUS (if we run the calculation with 14 cpus, then every group of 2 cpus will work on two replicas). The initial guess for the reaction path is given through 14 geometry files.
SCF_GUESS RESTART in combination with EXTRAPOLATION USE_GUESS has to be declared in the FORCE_EVAL/DFT section to allow for an efficient guess of the wavefunctions at each step of the BAND optimization.
Exercise 2: Metadynamics
The files for this part of the exercise are found in the EX1 folder.
Part 1
Obtain two restart files for independent configurations of the system at the geometry of the initial state thermalized at 450 K.
In the directory EQUIL the input file inp is used to run 3000 MD steps of MD in the NVT ensamble with a CSVR thermostat (a longer simulation should be used) two restart files obtained during the simulation (EQUIL-1_2000.restart and EQUIL-1_3000.restart) will be used to run a Metadynamics simulation with two WALKERS.
The trajectory of the MD should be also used to define the shape of the gaussians added in the metadynamics run.
Trajectories are printed in dcd format (EQUIL-pos-1.dcd use, e.g. vmd s.xyz EQUIL-pos-1.dcd to visualize the trajectory).
The MOTION section will look like:
&MOTION
&MD
ENSEMBLE NVT
STEPS 3000
TIMESTEP 0.5
TEMPERATURE 450.0
&THERMOSTAT
TYPE CSVR
REGION MASSIVE
&CSVR
TIMECON [fs] 200.0
&END
&END
&END MD
&PRINT
&TRAJECTORY
FORMAT DCD
&EACH
MD 100
&END
&END
&RESTART
&EACH
MD 1000
&END
FILENAME EQUIL
&END
&RESTART_HISTORY
&EACH
MD 1000
&END
&END
&END
&END
Part 2
The two restart files are copied in the directory Files (r1 and r2 respectively).
In the directory also the geometry for the whole system in the initial configuration as well as the geometry of the molecular fragment are included (files s and f respectively).
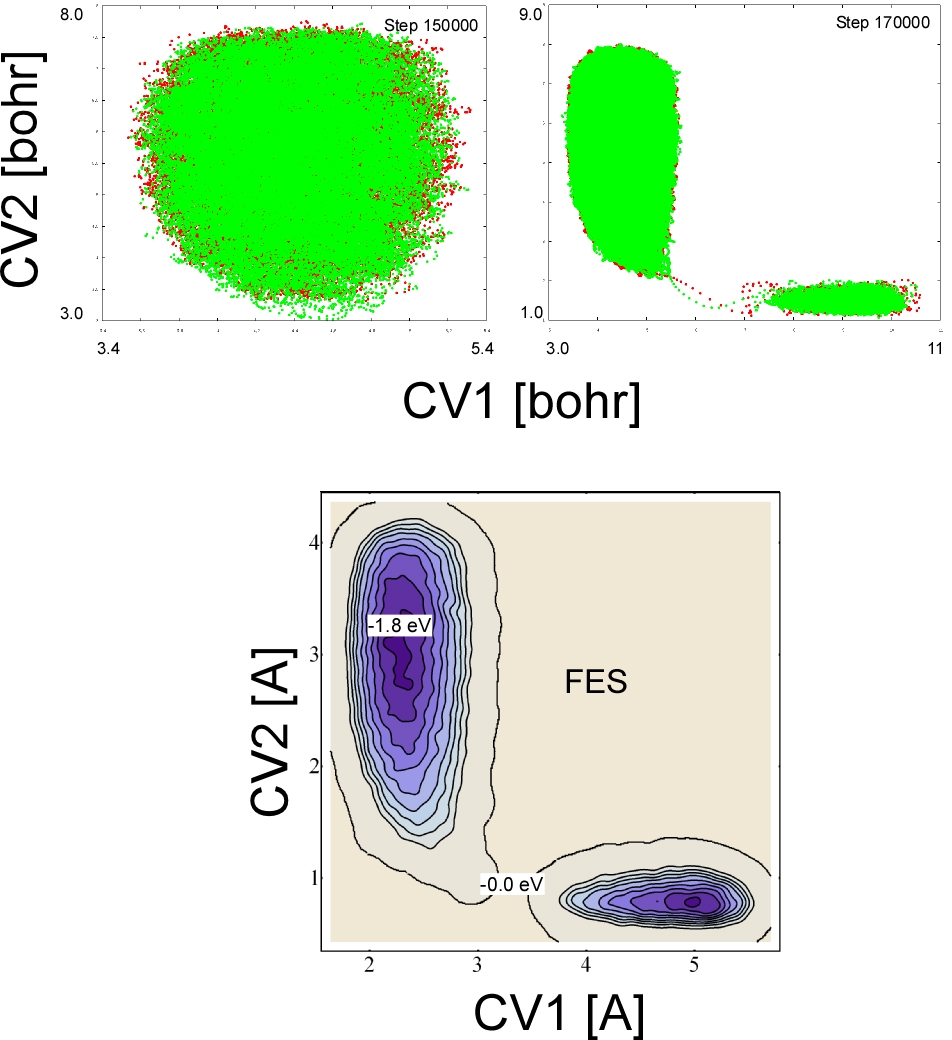
We perform a metadynamics simulation with two collective variables: the sum of C-H distances (CV1) and the H-H distance (CV2). Walls have to be introduced to limit the range of variability of the two CVs.
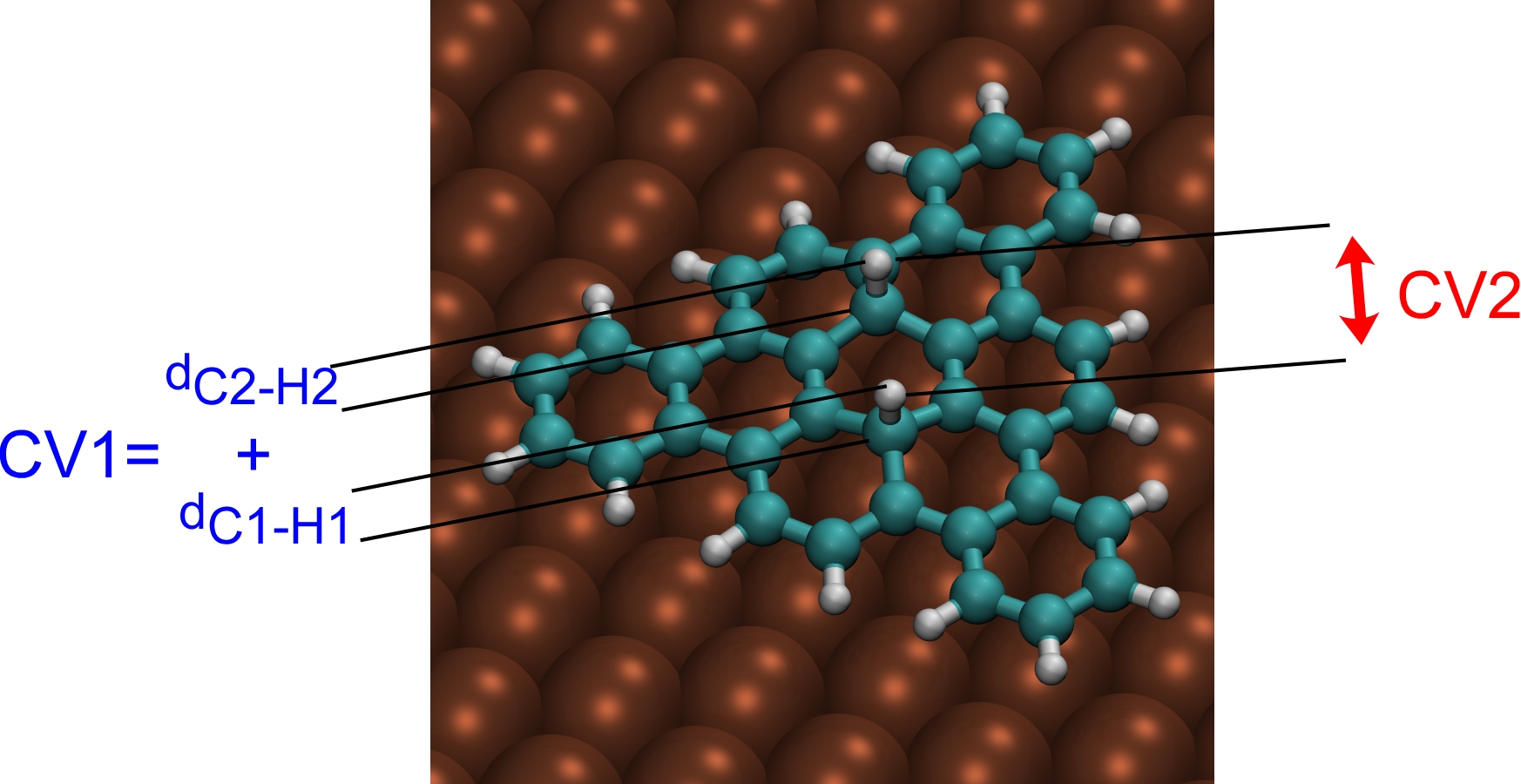
To define the collective variables introduce a COLVAR card in the SUBSYS section of the first FORCE_EVAL:
&SUBSYS
&COLVAR
&COMBINE_COLVAR
&COLVAR
&DISTANCE
ATOMS 4 50
&END
&END COLVAR
&COLVAR
&DISTANCE
ATOMS 7 49
&END
&END COLVAR
FUNCTION CV1+CV2
VARIABLES CV1 CV2
ERROR_LIMIT 1.0E-8
&END
&END
&COLVAR
&DISTANCE
ATOMS 49 50
&END
&END
The MOTION section has to be modified as follows to activate metadynamics on the two CVs:
&MOTION
&MD
ENSEMBLE NVT
STEPS 50000
TIMESTEP 0.5
TEMPERATURE 450.0
&THERMOSTAT
TYPE CSVR
REGION MASSIVE
&CSVR
TIMECON [fs] 200.0
&END
&END
&END MD
&FREE_ENERGY
&METADYN
DO_HILLS
NT_HILLS 50
WW 2.0e-3
&METAVAR
SCALE 0.1
COLVAR 1
&WALL
TYPE QUADRATIC
POSITION [angstrom] 5
&QUADRATIC
DIRECTION WALL_PLUS
K [kcalmol] 40.0
&END
&END
&WALL
TYPE QUADRATIC
POSITION [angstrom] 1.90
&QUADRATIC
DIRECTION WALL_MINUS
K [kcalmol] 40.0
&END
&END
&END METAVAR
&METAVAR
SCALE 0.1
COLVAR 2
&WALL
TYPE QUADRATIC
POSITION [angstrom] 3.80
&QUADRATIC
DIRECTION WALL_PLUS
K [kcalmol] 40.0
&END
&END
&WALL
TYPE QUADRATIC
POSITION [angstrom] 0.75
&QUADRATIC
DIRECTION WALL_MINUS
K [kcalmol] 40.0
&END
&END
&END METAVAR
# &METAVAR
# SCALE 0.1
# COLVAR 3
# &WALL
# TYPE QUADRATIC
# POSITION [angstrom] 4.00
# &QUADRATIC
# DIRECTION WALL_PLUS
# K [kcalmol] 40.0
# &END
# &END
# &WALL
# TYPE QUADRATIC
# POSITION [angstrom] 1.30
# &QUADRATIC
# DIRECTION WALL_MINUS
# K [kcalmol] 40.0
# &END
# &END
# &END METAVAR
&PRINT
&COLVAR
COMMON_ITERATION_LEVELS 4
&END
&END
&MULTIPLE_WALKERS
NUMBER_OF_WALKERS 2
WALKER_ID 1
WALKER_COMM_FREQUENCY 4
&WALKERS_FILE_NAME
../WALK_DATA_FILES/WALKER_1.data
../WALK_DATA_FILES/WALKER_2.data
&END
&END
&END METADYN
&END
&PRINT
&TRAJECTORY
FORMAT DCD
&EACH
MD 100
&END
&END
&RESTART
&EACH
MD 100
&END
FILENAME META
&END
&VELOCITIES ON
&EACH
MD 5000
&END
&END
&RESTART_HISTORY OFF
&END
&END
&END
&EXT_RESTART
RESTART_FILE_NAME ../../Files/r1
&END
The section specific to the multiple walkers could be removed to run the simulation with a single walker:
&MULTIPLE_WALKERS
NUMBER_OF_WALKERS 2
WALKER_ID 1
WALKER_COMM_FREQUENCY 4
&WALKERS_FILE_NAME
../WALK_DATA_FILES/WALKER_1.data
../WALK_DATA_FILES/WALKER_2.data
&END
&END
Here we declare that we are using two walkers, that the input belongs to the walker with ID 1, that the data relative to the different walkers are stored in the files contained in the directory WALK_DATA_FILES and that the history of the gaussian hills deposited by each walker should be exchanged every 4 hills deposited.
We need two independent directories (WALK1 and WALK2 in the example) belonging to each WALKER, each directory will contain an input in which the WALKER ID is modified (1 and 2).
The calculation will run as a FARMING: we need a common input that specifies that we are running two cp2k problems:
The input file for the calculation (inp in the directory EX1) will be:
&GLOBAL
PROGRAM FARMING
RUN_TYPE NONE
&END GLOBAL
&FARMING
NGROUP 2
&JOB
DIRECTORY WALK1
INPUT_FILE_NAME inp
&END JOB
&JOB
DIRECTORY WALK2
INPUT_FILE_NAME inp
&END JOB
&END FARMING
We declare that the cpus used for the calculation belongs to two groups of calculations,
one job has to run in the directory WALK1 according to the prescriptions contained in the input file WALK1/inp
and a second job has to run in the directory WALK2 (using WALK2/inp).
If we run the calculation with 8 cpus, 4 will be devoted to the JOB in directory WALK1 and 4 to WALK2.
Each group of 10 cpus will be then partitioned according to partition instruction contained in each inp file (GROUP_PARTITION 1 3).
During execution you can monitor the evolution of the CVs of each walker from the files
META-COLVAR.metadynLog and META-COLVAR.metadynLog
e.g.
plot 'WALK1/META-COLVAR.metadynLog' u 2:3 pt 7.0 ps 1.0 t "",\
'WALK2/META-COLVAR.metadynLog' u 2:3 pt 7.0 ps 1.0 t ""
You can reconstruct the free energy profile from a restart file with the command:
fes.popt -cp2k -mathlab -file META-META-1.restart -ndim 2 -ndw 1 2 -out fes_12.dat