
Calculation of surface energies of Au
In this exercise, we will build three small slabs based on the following figures; the cell is an orthorombic cell with repetition along z.
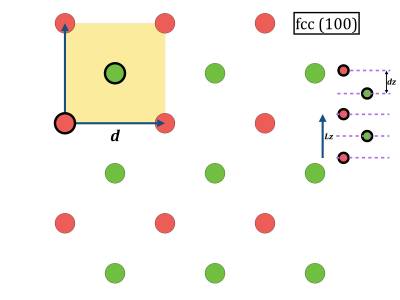


Figure out what is (in nearest neighbor units) the length of the three vectors Lx, Ly, Lz for the three high-symmetry surfaces.

Take the above table as reference for the orders of magnitude of surface energies.
- Download all the necessary files for computing the surface energies of all 3 high symmetry faces of gold from from the wiki: exercise_3.2.zip (all inputs are commented) in your home directory and unzip it:
you@eulerX ~$ wget http://www.cp2k.org/_media/exercises:2015_ethz_mmm:exercise_3.2.zip you@eulerX ~$ unzip exercises:2015_ethz_mmm:exercise_3.2.zip you@eulerX ~$ cd exercise_3.2
In principle all reconstructed phases should be with a lower surface energy, but this EAM potential fails for some of them. The input files are:
- 100_bulk.inp for determining the energy per atom in the bulk
- 100_unr.inp for the bulk termination (100)
- 100_rec.inp for the hexagonally reconstructed (100)
- 110_unr.inp for the (110) bulk termination
- 110_rec.inp for the (2×1) reconstruction
- 111.inp for the unreconstructed (111).
Before, however, to be able to run the cp2k program, we need to build the xyz files for all cases. First, we build the slabs, using this chain of commands (First assignment). Use for the following:
- (100) surface: use (in first neighbor units) LX LY LZ = ???????? ???????? 1.4142135 ; NX NY NZ = 4 5 5
- (110) surface: use (in first neighbor units) LX LY LZ = ???????? ???????? 1.0000000 ; NX NY NZ = 6 4 5
- (111) surface: use (in first neighbor units) LX LY LZ = ???????? ???????? 2.4494897 ; NX NY NZ = 4 3 3
- Find the correct values of lx ly for the three cases.
- use D = 2.885 (correct value for gold)
Example:
you@eulerxx $ m_lattice LX LY LZ NX NY NZ < 100.unit | m_xyzrescale D | m_xyzrefold 1 1 1 | m_xyzcenter 1 1 1 | m_xyzsort > 100_unr.xyz you@eulerxx $ m_lattice LX LY LZ NX NY NZ < 110.unit | m_xyzrescale D | m_xyzrefold 1 1 1 | m_xyzcenter 1 1 1 | m_xyzsort > 110_unr.xyz you@eulerxx $ m_lattice LX LY LZ NX NY NZ < 111.unit | m_xyzrescale D | m_xyzrefold 1 1 1 | m_xyzcenter 1 1 1 | m_xyzsort > 111_unr.xyz
Substitute the LX LY LZ (but ADD SOME VACUUM IN THE Z DIRECTION) in the ABC section of the *inp files. Check with vmd that the cell is ok. Run the cp2k code as
you@eulerxx $ bsub cp2k.popt -i 100_unr.inp -o 100_unr.out you@eulerxx $ bsub cp2k.popt -i 110_unr.inp -o 110_unr.out you@eulerxx $ bsub cp2k.popt -i 111_unr.inp -o 111_unr.out
The compute_surf* scripts give an idea how to compute the surface energies.
In the next step, we work also with 2 reconstructed surfaces: the 110_rec.xyz and the 100_rec.xyz.
you@eulerxx $ bsub cp2k.popt -i 100_rec.inp -o 100_rec.out you@eulerxx $ bsub cp2k.popt -i 110_rec.inp -o 110_rec.out
- Mark surface energies and relaxations of the first 2-3 top layers (use vmd or editor)
- Try to color the surfaces in vmd in a way that makes the height evident (ask the assistant) and produce snapshots (render)
- Comment on the (2×1) reconstruction: what do you see?
- Copy the 110_unr.xyz into a new file 110_my_rec.xyz
- Edit the file and remove one line each two on the two surfaces
- Check that the result is the same as in 110_rec.xyz
- Plot the same slabs, but adding periodic images in vmd: remember pbc set { a b c 90 90 90 }.