
Potential energy surface of alanine dipeptide
Alanine dipeptide is one of the simplest molecules that exhibits some important features common to larger biomolecules. In particular, it has more than one long-lived conformation, which we will identify in this exercise by mapping out its potential energy surface.
The carboxylate is replaced by an acyl group (C-terminus) and the primary amine is converted into a secondary amine by the addition of a methyl group (N-terminus).
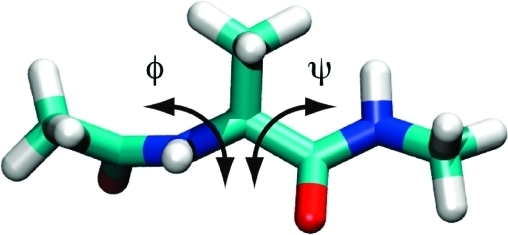
The conformations of alanine dipeptide are characterized by the dihedral angles of the backbone. Below, we color carbons in green, hydrogens in white, oxygen in red and nitrogen in blue, i.e. the torsional angle $\phi$ is C-N-C-C, while $\psi$ is N-C-C-N along the backbone.
a1a2ene.pdb with VMD and determine the atomic indices of the atoms defining the dihedral angles.
With this knowledge at hand, we will fix the dihedral angles and perform geometry optimization for all remaining degrees of freedom.
- The atomic indices defining the dihedral indices in the input file
geo.inare missing. ReplaceI1toI4by the atomic indices determined previously. Note: While VMD starts counting atoms from 0, CP2K starts counting from 1, i.e. the VMD indices need to be increased by 1. - Use
perform-gopt.shto perform the grid of geometry optimizations. - Use gnuplot to plot the potential energy surface (we have provided a script
epot.gp). Which are the two most favoured conformations? - Compare with Figure 3 of 10.1073/pnas.100127697.