
Ramachandran plot for Alanine Dipeptide
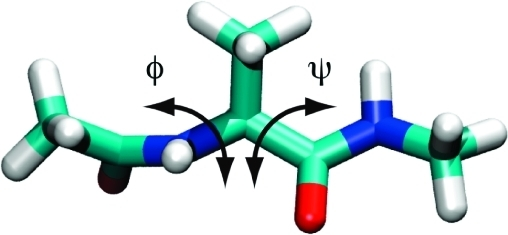
Alanine dipeptide is often studied in theoretical work because it is among the simplest systems to exhibit some of the important features common to biomolecules. It has more than one long-lived conformational state. The relevant angles are the dihedral angles of the backbone, commonly called Φ and Ψ (see figure). In the following scheme, light blue atoms are carbons, white ones are hydrogens, red are oxygens, and blue are nitrogens. So the torsional angle Φ is C-N-C-C and Ψ is N-C-C-N along the backbone.
A detailed study of this system (see 10.1073/pnas.100127697) shows the presence, in vacuum of two stable states:
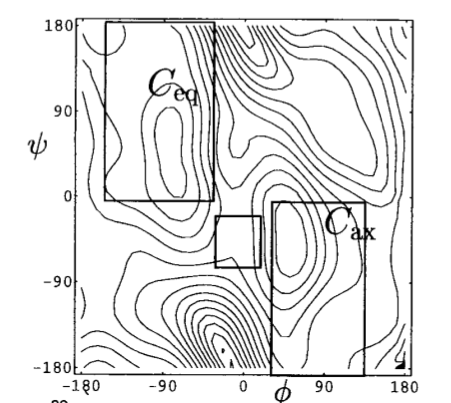
The diagram represents the contour lines of the molecular energy as a function of the two dihedral angles. Contour lines are separated by 2 kcal/mol.
In this exercise you will obtain a simplified version of the above potential energy surface, obtained in a very similar way as in the paper. You will constrain the angles at fixed values using a strong harmonic potential, and optimize all other degrees of freedom. From this, a grid of energies will be built, and the energy diagram (Ramachandran plot) will be constructed.
Download the 2.2 exercise into your $HOME folder and unzip it.
you@eulerX ~$ wget http://www.cp2k.org/_media/exercises:2015_ethz_mmm:exercise_2.2.zip you@eulerX ~$ unzip exercises:2015_ethz_mmm:exercise_2.2.zip
Then go to the directory “exercise_2.2/”
you@eulerX ~$ cd exercise_2.2
The input file has a section concerning restrained optimization:
- inp.templ
&FORCE_EVAL ! This section defines method for calculating energy and forces METHOD FIST ! Using Molecular Mechanics &MM &FORCEFIELD ! This section specifies forcefield parameters parm_file_name ace_ala_nme.pot ! This file contains force field parameters parmtype CHM ! forcefield parameters has CHARMM format &SPLINE ! This section specifies parameters to set up the splines used ! in the nonboned interactions (both pair body potential and many body potential) EMAX_SPLINE 1.0 ! Specify the maximum value of the potential up to which splines will be constructed &END &END FORCEFIELD &POISSON ! This section specifies parameters for the Poisson solver &EWALD ! This section specifies parameters for the EWALD summation method (for the electrostatics) EWALD_TYPE ewald ! Standard non-fft based ewald method ALPHA .36 GMAX 29 ! Number of grid points &END EWALD &END POISSON &PRINT ! This section controls printing options &FF_INFO &END &FF_PARAMETER_FILE &END &END &END MM &SUBSYS ! This section defines the system &CELL ! Unit cell set up ABC 50.0 50.0 50.0 ! Lengths of the cell vectors A, B, and C &END CELL &COLVAR ! This section specifies collective varialbe &TORSION ! This section defines variable as torsion angle ATOMS 5 7 9 15 ! Four atoms specify torsion angle &END &PRINT &END &END &COLVAR ! This section specifies collective varialbe &TORSION ! This section defines variable as torsion angle ATOMS 7 9 15 17 ! Four atoms specify torsion angle &END &PRINT &END &END &TOPOLOGY CONN_FILE_NAME ace_ala_nme.psf ! File which contains the connectivity information CONNECTIVITY psf ! Format of the connectivity file is PSF COORD_FILE_NAME ini.pdb ! File which contains atom's coordinates of the system COORDINATE PDB ! Coordinates are in the PDB format &DUMP_PDB ! This sections specifies the dumping of the PDB at the starting geometry &END &DUMP_PSF ! This sections specifies the dumping of the PSF connectivity &END &END TOPOLOGY &PRINT &TOPOLOGY_INFO AMBER_INFO &END &END &END SUBSYS &END FORCE_EVAL &GLOBAL ! Section with general information regarding which kind of simu lation to perform an parameters for the whole PROGRAM PRINT_LEVEL LOW ! Global print level PROJECT ch ! Name of the project. This word will appear as part of a name of all ouput files (except main ouput file, specified with -o option) RUN_TYPE GEO_OPT ! Run type is a geometry optimization &END GLOBAL &MOTION ! This section specifies a set of tool connected with the motion of the nuclei &CONSTRAINT ! Section specifying information regarding how to impose constraints on the system &COLLECTIVE INTERMOLECULAR T COLVAR 1 ! Sequential number of the variable &RESTRAINT ! This section specifies how stong is the restraint K=5.0 ! U(x)=K*(x-x0)^2 &END TARGET [deg] _A1_ ! _A1_ will be changed to the number by an external scritp &END &COLLECTIVE INTERMOLECULAR T COLVAR 2 ! Sequential number of the variable &RESTRAINT ! This section specifies how stong is the restraint K=5.0 ! U(x)=K*(x-x0)^2 &END TARGET [deg] _A2_ ! _A1_ will be changed to the number by an external scritp &END &END &PRINT ! This section controls the printing properties during an optimization or MD run &TRAJECTORY FORMAT PDB ! Format of the ouput trajectory is PDB ADD_LAST NUMERIC ! (Wiki) If the last iteration should be added, and if it should be marked symbolically (with l) or with the iteration number.Not every iteration level is able to identify the last iteration early enough to be able to output. When this keyword is activated all iteration levels are checked for the last iteration step. &EACH GEO_OPT 100 ! Print one frame every 100 iteration &END &END &END &GEO_OPT ! This section specifies optimizer options OPTIMIZER BFGS ! Type of the optimizer MAX_ITER 5000 ! Maximum number of optimization steps MAX_FORCE 0.005 ! The value of maximal force RMS_FORCE 0.003 ! The value of maximal force RMS &END &END
At this point submit the job grid, first loading the module for cp2k version 2.5 entering
you@eulerX exercise_2.2$ module load cp2k you@eulerX exercise_2.2$ bsub < grid_alanine
The file “a1a2ene” will contain three columns: the two constrained dihedral angles, and the corresponding energy. Using gnuplot, you can visualize the results
you@eulerX exercise_2.2$ gnuplot torsion.gnu
you@eulerX exercise_2.2/Logs$ m_pdbtorsion 5 7 9 15 ; m_pdbtorsion 7 9 15 17
for the two dihedral Phi and Psi.
To clean the output and unnecessary files and do it again,
you@eulerX exercise_2.2$ ./cleanall