
Dehydration of ethanol
We will investigate today a very important chemical reaction, the production of ethene (ethylene) from ethanol. Ethanol is heated with an excess of concentrated sulphuric acid at a temperature of 170°C. The gases produced are passed through sodium hydroxide solution to remove the carbon dioxide and sulphur dioxide produced from side reactions. Another way to favor this reaction is in presence of a catalyst surface, which makes the reaction exothermic even at room temperature, and the free energy barrier for the reaction is highly reduced, so to obtain reasonable reaction rates. The use of hematite (iron-based nanocrystal) as a catalyst is described computationally in the following paper by Lopes et al: 10.1039/c6ra08509a. We are interested in the gas phase reaction, in particular in the reactants and the products energy, to estimate the “energy of reaction”.
The reaction is the following:
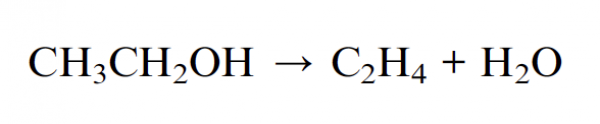 and this is the pictorial view in the gas phase:
and this is the pictorial view in the gas phase:
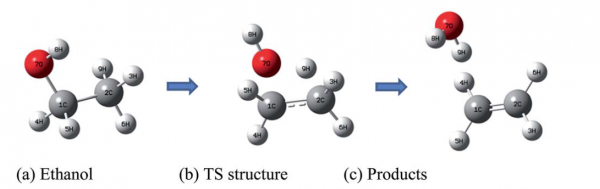
In the supplementary material of the paper, which can be found here, several calculation with different methods ranging from DFT to hybrid methods to high-level methods are described, together with the basis set dependence of the results.
We will compare our results with the published ones.
The input file structure is shown below:
&DFT
&POISSON ! Solver requested for non periodic calculations
PERIODIC NONE
PSOLVER WAVELET ! Type of solver
&END POISSON
&QS ! Parameters needed to set up the Quickstep framework
METHOD GAPW ! Method: gaussian and augmented plane waves
&END QS
#
# Include the exchange and correlation information
#
@INCLUDE './pbe.inc'
&END DFT
&SUBSYS ! This section defines the system
&CELL ! Unit cell set up
PERIODIC NONE ! Non periodic calculation
ABC 10 10 10 ! Lengths of the cell vectors A, B, and C
&END CELL
&COORD ! This section specify all the atoms and their coordinates
H 2.5558925119 4.9201502245 3.9999999733
H 2.5617214834 3.0599837703 4.0000000267
C 3.3235178526 3.9916405319 4.0000000002
C 4.6493228423 3.9951429630 3.9999999995
H 5.4118318595 4.9261779106 4.0000000269
H 5.4167134504 3.0669045998 3.9999999735
&END COORD
&TOPOLOGY
&CENTER_COORDINATES
&END
&END TOPOLOGY
&KIND H ! potential and basis for H
&BASIS
@INCLUDE './H_631Gdp.inc'
&END
POTENTIAL ALL
&POTENTIAL
1 0 0
0.20000000 0
&END
&END KIND
&KIND C ! potential and basis for C
&BASIS
@INCLUDE './C_631Gdp.inc'
&END
POTENTIAL ALL
&POTENTIAL
4 2 0
0.34883045 0
&END
&END KIND
&END SUBSYS
&END FORCE_EVAL ! This section defines method for calculating energy and forces
&GLOBAL ! Section with general information regarding which kind of simulation to perform an parameters for the whole PROGRAM
PRINT_LEVEL LOW ! Global print level
PROJECT c2h4 ! Name of the project. This word will appear as part of a name of all ouput files (except main ouput file, specified with -o option)
RUN_TYPE GEO_OPT ! Geometry optimization
&END GLOBAL
The exercise is executed on the hypatia cluster. The instructions to connect are found here. After creating a directory as usual, you can copy the files from the following location:
Copy the files to the created directory in the /mnt/project/yourusername/ path.
h2o.inpfor the geometry optimization of waterethanol.inpfor the geometry optimization of ethanolethylene.inpfor the geometry optimization of ethylene (ethene)runto launch the jobs to the queue- several
*.incfile describing the level of theory (exchange and/or correlation functional) and the basis sets.
The command to launch the job is
where file.inp has to be replaced by the relevant prefix of the input file (example: h2o).
In the *.xyz files you can look for the final energies by the following command:
. This will list all the energies. Pick the last one for the optimized structure. You can also visualize it with vmd.
You will run the calculation by changing in ALL THREE INPUT FILES the @INCLUDE lines, in the following way:
At the line concerning the exchange and correlation potential, you prescribe the level of theory:
- hf.inc for Hartree-Fock
- pbe.inc for DFT/PBE, with gradient corrected local density
- b3lyp.inp for hybrid functional containing a percentage of exact exchange
When you are done with the three levels of theory, then you can redo the exercise with the larger basis set, and change EVERYWHERE in the input file the instances of H_631Gdp.inc to H_6311++G2d2p.inc and the same for the O_* and C_* instances.
Redo the calculations with the three levels pbe, b3lyp and hf.
- Compute the reaction energy for the dehydration reaction of ethanol
- Prepare a table with rows and columns: on the rows the level of theory, on the columns the basis set (3×2 table)
- Compare the results with the published ones (note the conversion factors. You can use the tool at Energy converter
- Comment on the dependence on the basis set and on the level of theory (hint: this also need the next theory lecture)
- Is this information enough to determine the rates of reaction? Why?
BONUS TRACK
ethanol.inp into ethanol_dens.inp.
Add the following sections: under &DFT
&PRINT
&E_DENSITY_CUBE
&END
&END
&SCF
SCF_GUESS RESTART
&END
This tells to read the old wavefunction and to print the cubefile of the density.
At the end of the input file:
&EXT_RESTART
RESTART_FILE_NAME ./c2h6o-1.restart
&END
Then, change RUN_TYPE GEO_OPT to RUN_TYPE ENERGY to only run a single point calculation. It will generate a cubefile with the density which you may visualize with VMD.